In August 2021, Amol Akhade, an oncologist at Nair Medical Hospital in Mumbai, India, received an e-mail from the Swiss drug manufacturer Roche recommending the use of a drug named atezolizumab to treat a specific kind of breast cancer. Akhade was surprised. That month, Roche had withdrawn the drug for this purpose in the United States (although it is still approved to treat other kinds of cancer).
For the type of breast cancer in question — known as triple-negative because it lacks three key protein markers — atezolizumab was made available in 2019 through the US Food and Drug Administration’s (FDA’s) Accelerated Approval Program. Accelerated approval is a fast-track process designed to place desperately needed drugs in the hands of patients quicker than is possible with conventional drug approval. But a follow-up study found that atezolizumab made little difference to tumour growth, and that people who received it were less likely to survive up to two years after treatment than were those not taking it 1 . When the FDA received these data in 2021, it indicated that accelerated approval was no longer appropriate, and Roche withdrew the drug for this form of breast cancer. The same thing happened with the European Medicines Agency (EMA), based in Amsterdam, but not everywhere else.
In India, for example, where the drug was still approved for triple-negative breast cancer, Roche continued to promote the treatment until at least September, a fact that Akhade says he found “quite shocking”. “It’s not like patients with triple-negative breast cancer are different in the United States and outside,” he says. “We cannot have such differential treatment.” When asked about its responsibility to patients, Genentech, the Roche subsidiary in San Francisco, California, that developed atezolizumab, said that the drug is still approved in 100 countries for triple-negative breast cancer. “All of our medicines strictly adhere to the regulatory and promotional requirements of all local healthcare authorities,” said an e-mail from the company.
Accelerated approvals in the United States are granted on the basis of clinical studies that suggest a health benefit without necessarily demonstrating it fully. The process prioritizes speed over certainty, and it requires companies to complete follow-up studies to confirm a treatment’s benefits. This is a “very reasonable compromise”, says bioethicist Holly Fernandez Lynch at the University of Pennsylvania in Philadelphia, “so long as we can get the confirmatory evidence quickly and reliably”.
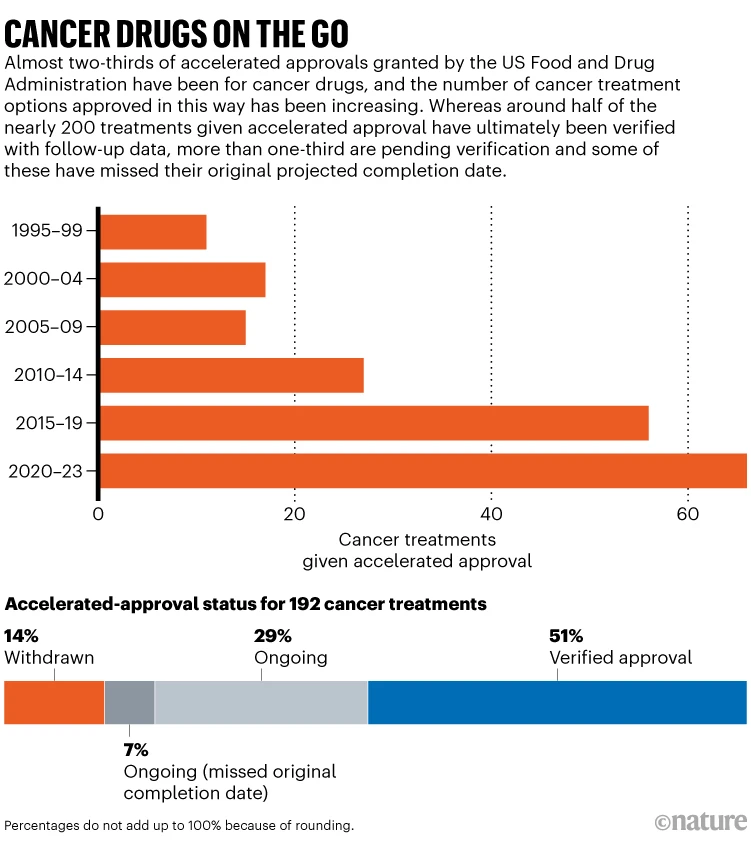
But there are complications that have made the programme controversial. Companies don’t always conduct studies in a timely manner, and the FDA hasn’t always been great about enforcing them. Researchers have noted that the median time to withdrawal for a drug that fails to hold up in confirmatory studies is four years 2 ; sometimes it takes decades. And some high-profile accelerated approvals have raised eyebrows. Experts worry that the evidence used to support the approval of the Alzheimer’s disease drug aducanumab was insufficient; three FDA advisers resigned in protest. Other specialists have complained that the process allows companies to market expensive drugs for rare childhood diseases to desperate parents without sufficient evidence. There are currently nearly 200 cancer treatments approved through this pathway. Follow-up studies have led to the withdrawal of 26, and 68 are still awaiting confirmatory evidence (see ‘Cancer drugs on the go’).
A lot of prescriptions can be made in the time between a drug’s accelerated approval and the publication of follow-up results. If those results are negative, the drug can still be prescribed to many people before its eventual withdrawal, and even afterwards — particularly when professional medical bodies recommend its use. And accelerated approvals have an effect beyond US borders, thanks to the fact that many countries use FDA decisions to guide their own regulatory policies.
At the heart of the problem, says Lynch, is a failure of communication — both in the outcomes of follow-up studies and the nature of accelerated approval itself. “Clinicians and patients often view FDA approval as an on–off switch — either a drug is approved or it’s not,” she says. But it’s not that simple.
Communication gaps
The Accelerated Approval Program was established in 1992, as a result of efforts from activists and advocates during the HIV epidemic in the 1980s. At the time, people facing a deadly disease were willing to accept a little uncertainty in exchange for quicker access. It allowed drug makers to use a ‘surrogate’ endpoint to gauge a treatment’s purported benefits. For example, studies of most cancer drugs estimate whether a treatment can pause a tumour’s growth — progression-free survival — rather than looking at metrics such as tumour regression or extended lifespan. Although this endpoint is thought to correlate to a longer life, evidence for this link is mixed. 3
The distinction between surrogate endpoints and more direct ones isn’t always obvious, even to clinicians, says Reshma Ramachandran, a health-services researcher at Yale University in New Haven, Connecticut. “Manufacturers have made it seem that accelerated approval is just like any other approval. It’s not, and that level of uncertainty is not really being translated to clinicians,” she says.
Ravi Parikh, a medical oncologist at the University of Pennsylvania, often navigates the maze of evidence for the use of different drugs. He parses data from clinical trials as well as guidance offered by groups such as the National Comprehensive Cancer Network (NCCN), a non-profit alliance of US clinical cancer centres that helps to create guidelines for practitioners, based in Plymouth Meeting, Pennsylvania. Parikh has seen accelerated-approval drugs help his patients to live longer. But he has sometimes felt unsure about the benefits of a newly available drug for a person in need. “There’s a temptation to use them even though they may not be as effective,” he says.
To understand how others deal with that inclination, Parikh and his colleagues reviewed how doctors responded to the accelerated approval of five cancer medications that were later withdrawn for lack of confirmatory evidence 2 They found that clinicians quickly reached for the treatments when they became available. About one-quarter of eligible people received an accelerated-approval drug. “The highest period of utilization for most of these drugs was within the first year after they were approved,” Parikh says. When further evidence failed to confirm a drug’s effectiveness, usage began to slow but did not stop. Clinicians continued to prescribe accelerated-approval drugs to 10% of eligible people, even after the drugs had been withdrawn (see ‘Treatment updates’).
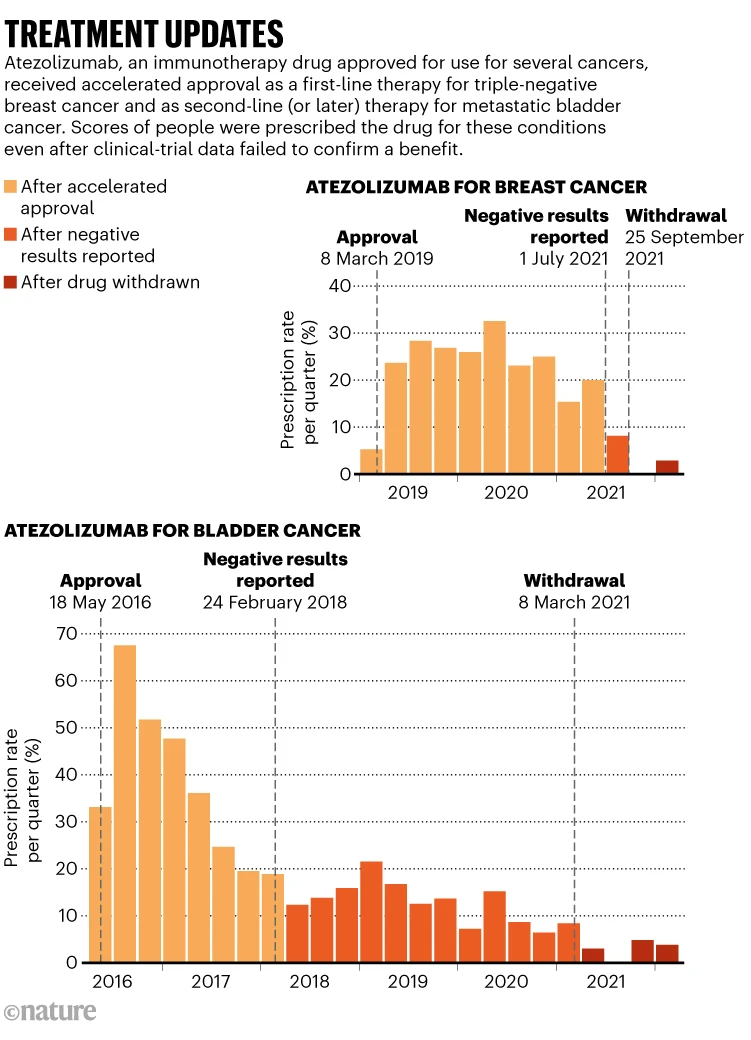
For some bladder cancers, immunotherapy drugs that received accelerated approval were later found to be less effective than were older chemotherapies. “In many cases, the previous standard of care is better,” Parikh says.
And if they’re on par with the existing options, Parikh says that clinicians must consider both the cost, which is often substantially higher for newer drugs, and the differences in side effects. “For that reason, there’s still maybe some downsides of using it, even if it’s not substantially worse.”
As for why doctors prescribe drugs after they’ve been withdrawn, Parikh chalks it up to habit. “Doctors get used to prescribing a drug,” he says. “It takes time to change.”
Guidelines offered to clinicians by the NCCN can strengthen those habits — not only because clinicians rely on them for treatment plans, but also because this guidance determines federal health-care reimbursement. Knowing that patients will be reimbursed for specific drugs influences clinicians’ choices, Ramachandran says.
In an analysis of 36 drugs that were granted accelerated approval between 2016 and 2018, Ramachandran and her colleagues found inconsistencies in how the NCCN’s guidelines were updated. If the FDA had converted a drug to full conventional approval on the basis of strong confirmatory trials, the NCCN guidance often reflected that decision. But the recommendation was less likely to be updated after an FDA withdrawal 4
The guidelines are regularly updated “to always reflect the current therapeutic landscape and FDA approval status”, an NCCN spokesperson said in an e-mail statement to Nature.
Only recently have clinicians grown more aware that accelerated-approval drugs might rely on murky evidence, and that’s a result of efforts from advocacy groups such as Doctors for America, Ramachandran says.
Global impact
The effects of accelerated approval can spread beyond US borders. Regulatory agencies in low- and middle-income countries sometimes rely on decisions made by the FDA and EMA. Some lack their own regulatory framework and so follow the FDA’s decisions directly, under the guidance of the World Health Organization. “Particularly in Latin America, what the FDA says dictates a lot of the things that our regulatory authorities do,” says Enrique Soto, an oncologist at the National Institute of Medical Sciences and Nutrition in Mexico City. In part owing to long regulatory delays during the COVID-19 pandemic, drugs approved by the FDA and EMA were allowed to be considered for use, even without regulatory approval from Mexican authorities, Soto says.
For India, which has its own regulatory body, the Central Drugs Standard Control Organisation (CDSCO), and others, the position that the FDA takes on a drug can be very influential. “The FDA is still seen as the gold standard in terms of regulatory review and approval,” Ramachandran says. “They play a huge role in the international space in terms of setting the bar for what sorts of evidence should be accepted.”
But, Soto says, nuances of precisely which FDA approval pathway was used or what surrogate endpoints were measured are often lost in the process. “The concept of accelerated approval is poorly known outside of the United States,” Soto says.
In an e-mail statement, an FDA spokesperson said, “The FDA does not regulate drug products intended for markets outside of the United States. However, the agency encourages global regulatory cooperation and harmonization to improve efficiency of drug development and post-marketing safety surveillance.”
In the case of atezolizumab for triple-negative breast cancer, the CDSCO permitted the drug’s use after the FDA granted accelerated approval in March 2019, but did not mirror the FDA’s subsequent actions.
In a February 2022 commentary 5 , Akhade and his colleagues reported on how this happened for atezolizumab and nine other accelerated-approval drugs that were withdrawn in the United States the previous year. Although manufacturers used the accelerated approvals to promote the medicines in low- and middle-income countries, they did not withdraw them there when the data failed to support their continued use. “Companies should be accountable to patients across the globe,” Akhade says. “It’s a moral responsibility.”
Following discussions between the drugmaker and the CDSCO in June 2022, atezolizumab remained approved for triple-negative breast cancer. “All of our medicines strictly adhere to the regulatory and promotional requirements of all local health-care authorities,” a Genentech spokesperson said in an e-mail to Nature. The CDSCO did not respond to requests for comment.
Medical oncologist Bishal Gyawali at Queen’s University in Kingston, Canada, who co-authored the commentary with Akhade, says that it’s time for international drug regulators to look beyond the FDA and EMA for guidance. Different countries experience different rates of certain cancers, and low- and middle-income countries should prioritize drugs for those that are most relevant to their populations instead of relying on the FDA “as a surrogate”, he says.
But Matthew Herder, a pharmaceutical-law researcher at Dalhousie University in Halifax, Canada, says that the FDA remains an international standard, because regulators around the world don’t have equivalent resources. As a result, the responsibility to protect people with cancer globally needs to be shared, he says. “The WHO needs to take more seriously the problems with established systems like the FDA,” he says. “We need a more honest conversation about harmonizing.”
Looking ahead
Reforms are possible — at least in the United States. In late 2022, the US government enacted the Food and Drug Omnibus Reform Act (FDORA), which aims to make the accelerated-approval process more transparent and gives the FDA greater authority to ensure that drug makers complete confirmatory studies.
The FDA now requires companies to begin a clinical trial to gather the data needed for a conventional approval before they apply for accelerated approval. It has also begun re-evaluating previous accelerated approvals, a process that has led to high-profile withdrawals. Other safeguards give the FDA more authority to ensure that companies withdraw drugs when required to do so. “FDORA did a number of good things in terms of increasing the FDA’s oversight,” Ramachandran says. But the long-term benefits hinge on its implementation, she adds, particularly of the FDA’s ability “to play more of a role in making clear not just to manufacturers, but also to patients and clinicians, what the level of evidence is at the time of approval”.
Parikh says that the Accelerated Approval Program could be strengthened further by gathering more data during preliminary studies. For instance, investigators could track whether the treatment helps people to survive longer than is typically observed with standard clinical treatments. Comparisons of a drug in a clinical study with real-world data are imperfect, Parikh says, but could still help to guide decision-making. And, Ramachandran says, professional guidelines that more accurately reflect the FDA’s decisions could reduce potential harms. “It’s very low-hanging fruit for the guidelines to explicitly state when a drug has been through the accelerated-approval pathway.”
A spokesperson for the NCCN said that details of the evidence backing the use of a drug are not in the guidelines, but are included in other NCCN resources for clinicians.
Spreading that message internationally could also help regulatory authorities in low- and middle-income countries to weigh up the pros and cons of accelerated-approval drugs when making decisions. Herder says that the uncertainty that accompanies accelerated-approval drugs makes them “by far the riskiest drugs available”. Until that message spreads, he adds, regulators in low- and middle-income countries relying on the FDA’s accelerated-approval decisions might do “more harm than good”.
doi: https://doi.org/10.1038/d41586-023-02492-x
References
- Miles, D. et al. Ann. Oncol. 32, 994–1004 (2021).
- Parikh, R. B. et al. JAMA Oncol. 9, 567–569 (2023).
- Belin, L., Tan, A., De Rycke, Y & Dechartres, A. Br. J. Cancer 122, 1707–1714 (2020).
- Mooghali, M. et al. J. Clin. Oncol. 41, 1590 (2023).
- Akhade, A., Sirohi, B. & Gyawali, B. Lancet Oncol. 23, 201–203 (2022).
Jyoti Madhusoodanan, a 2022 APF fellow, is examining the policies and practices behind clinical trials.